Clinical Trials and the FDA
Before a new drug/treatment, clinical device or procedure can be widely used by doctors to treat patients, these potential medical tools undergo (usually very long) clinical trials. These trials use volunteers to evaluate efficacy (how well the treatment works) and most importantly, safety (side effects). Before anything gets OKed by the FDA (Food and Drug Administration) in the US, it has to be shown to be effective and safe via clinical trials results, which get reported to the FDA, reviewed by experts and eventually given an official approval.
I remember times when “clinical trials” or “FDA approval” were not common terms, but now you may hear in the health news that a new drug seems promising for a specific condition (diabetes, blood pressure, cholesterol, Alzheimer’s, Parkinson’s, etc) with a warning that this potential treatment will not be available to patients until it has gone through many additional trials (up to 10 years depending on the treatment and the target condition).
In some cases when the drug/treatment is being tested at the NIH or different medical centers, terminal patients may be admitted as volunteers in the trial - you may know people/family who have been in such trials (or may be interested in being a volunteer on a clinical trial yourself), especially when they have received approved treatments that failed for cancer for example. The free database ClinicalTrials.gov includes clinical studies worldwide (currently a total of 293,889 research studies in all 50 states and in 207 countries) and is searchable by condition or disease, drug name, and country at:
https://clinicaltrials.gov/
All trials have to obtain approval by relevant committees and institutional review boards that make sure ethical requisites are being met, such as “informed consent” by the volunteers in the trial, which is requested prior to the trial. Volunteers have to be informed of the treatment being tested, possible side effects and risks, aims of the trial, after which they give consent to participating as volunteers and also to what can be done with the information and materials (that may be stored for years, such as blood samples) obtained from the trial; everything is of course kept confidential even if pooled results can be presented and shared with other/relevant audiences. Al clinical trials are expected to comply with "good clinical practice" (GCP) guidelines, which ensure ethical and scientific trials and protect human subjects' rights and confidentiality. Recruitment, selection and final inclusion of candidate volunteers into a clinical trial is a process that may take several months in order to reach the required numbers that will allow statistical analysis for the treatment to be proven effective and safe.
Pre-clinical Research Phase
Before clinical trials start, there has to be evidence (data) that the drug or test/device to be trialed is promising in preliminary laboratory studies done in cells in culture and/or animal models (usually mice) for the specific disease or condition (see my post on animal models). These “pre-clinical” studies results, along with detailed explanation of procedures by which the drug to be tested is made in batches and specific “protocols” that will be followed in the clinical trial proposed including following ethics rules, are submitted to the FDA in what is known as a “investigational new drug” or IND application; all clinical trial phases thereafter are under FDA oversight. Clinical trial phases 1-4 occur consecutively, and FDA approval to move on to the next phase depends on results and analysis of the previous phase, which are timely submitted to the FDA for review.
Before a new drug/treatment, clinical device or procedure can be widely used by doctors to treat patients, these potential medical tools undergo (usually very long) clinical trials. These trials use volunteers to evaluate efficacy (how well the treatment works) and most importantly, safety (side effects). Before anything gets OKed by the FDA (Food and Drug Administration) in the US, it has to be shown to be effective and safe via clinical trials results, which get reported to the FDA, reviewed by experts and eventually given an official approval.
I remember times when “clinical trials” or “FDA approval” were not common terms, but now you may hear in the health news that a new drug seems promising for a specific condition (diabetes, blood pressure, cholesterol, Alzheimer’s, Parkinson’s, etc) with a warning that this potential treatment will not be available to patients until it has gone through many additional trials (up to 10 years depending on the treatment and the target condition).
In some cases when the drug/treatment is being tested at the NIH or different medical centers, terminal patients may be admitted as volunteers in the trial - you may know people/family who have been in such trials (or may be interested in being a volunteer on a clinical trial yourself), especially when they have received approved treatments that failed for cancer for example. The free database ClinicalTrials.gov includes clinical studies worldwide (currently a total of 293,889 research studies in all 50 states and in 207 countries) and is searchable by condition or disease, drug name, and country at:
https://clinicaltrials.gov/
All trials have to obtain approval by relevant committees and institutional review boards that make sure ethical requisites are being met, such as “informed consent” by the volunteers in the trial, which is requested prior to the trial. Volunteers have to be informed of the treatment being tested, possible side effects and risks, aims of the trial, after which they give consent to participating as volunteers and also to what can be done with the information and materials (that may be stored for years, such as blood samples) obtained from the trial; everything is of course kept confidential even if pooled results can be presented and shared with other/relevant audiences. Al clinical trials are expected to comply with "good clinical practice" (GCP) guidelines, which ensure ethical and scientific trials and protect human subjects' rights and confidentiality. Recruitment, selection and final inclusion of candidate volunteers into a clinical trial is a process that may take several months in order to reach the required numbers that will allow statistical analysis for the treatment to be proven effective and safe.
Pre-clinical Research Phase
Before clinical trials start, there has to be evidence (data) that the drug or test/device to be trialed is promising in preliminary laboratory studies done in cells in culture and/or animal models (usually mice) for the specific disease or condition (see my post on animal models). These “pre-clinical” studies results, along with detailed explanation of procedures by which the drug to be tested is made in batches and specific “protocols” that will be followed in the clinical trial proposed including following ethics rules, are submitted to the FDA in what is known as a “investigational new drug” or IND application; all clinical trial phases thereafter are under FDA oversight. Clinical trial phases 1-4 occur consecutively, and FDA approval to move on to the next phase depends on results and analysis of the previous phase, which are timely submitted to the FDA for review.
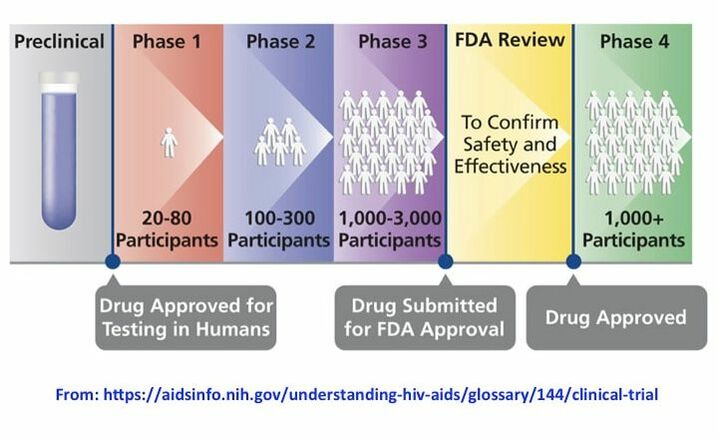
Clinical Trial Phases
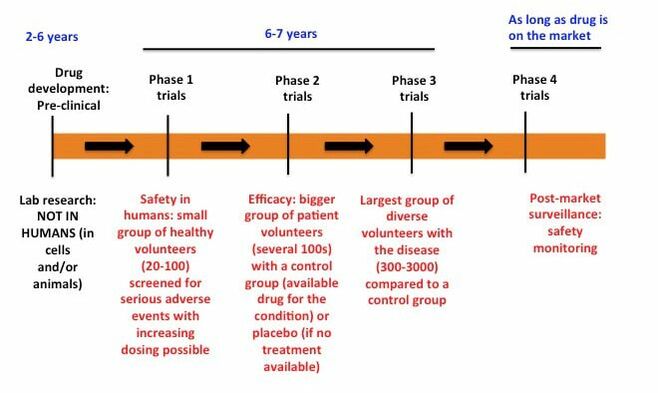
There are 4 phases of clinical trials which may happen consecutively, with progress to the next phase depending on approval of previous phase results, or sometimes two phases may happen simultaneously depending on trial design. Planning and execution of each phase require time and navigating loads of associated paperwork, administrative and regulatory requirements, with possible delays affecting progress. Trials may be conducted at different sites sometimes in different countries; in these cases procedures need to be similar so results are comparable, and forms for volunteers are provided in the local language.
The first phase (phase 1) usually recruits a small number of healthy volunteers to look at dosing of the drug and side effects, sometimes including studies on how/when the drug is processed (metabolized) and stays in the body. Phases 2 and 3 involve larger groups of volunteers including patients with the disease/condition and “control” groups to compare with, who receive either an existing treatment or a “placebo” (dummy treatment that looks exactly like the real treatment). There are instances in which trials may be sped up or skip phases depending on previous testing. For example, a “repurposed” drug that has been tested for one disease/condition and may be considered for another usually is accelerated to phase 2, as its safety in humans was preciously tested in phase 1 trials for the first condition (already FDA-approved). A "new drug application" (NDA) is submitted to the FDA after phase 3 is completed to demonstrate that the drug is safe and effective for its intended use in the population studied; the NDA is a complete and extensive report including pre-clinical and phases 1-3 data.
There are 4 phases of clinical trials which may happen consecutively, with progress to the next phase depending on approval of previous phase results, or sometimes two phases may happen simultaneously depending on trial design. Planning and execution of each phase require time and navigating loads of associated paperwork, administrative and regulatory requirements, with possible delays affecting progress. Trials may be conducted at different sites sometimes in different countries; in these cases procedures need to be similar so results are comparable, and forms for volunteers are provided in the local language.
The first phase (phase 1) usually recruits a small number of healthy volunteers to look at dosing of the drug and side effects, sometimes including studies on how/when the drug is processed (metabolized) and stays in the body. Phases 2 and 3 involve larger groups of volunteers including patients with the disease/condition and “control” groups to compare with, who receive either an existing treatment or a “placebo” (dummy treatment that looks exactly like the real treatment). There are instances in which trials may be sped up or skip phases depending on previous testing. For example, a “repurposed” drug that has been tested for one disease/condition and may be considered for another usually is accelerated to phase 2, as its safety in humans was preciously tested in phase 1 trials for the first condition (already FDA-approved). A "new drug application" (NDA) is submitted to the FDA after phase 3 is completed to demonstrate that the drug is safe and effective for its intended use in the population studied; the NDA is a complete and extensive report including pre-clinical and phases 1-3 data.
Phase 2 and 3 trials may be “randomized” and/or “blind”. Randomization is usually computer-based, assigning volunteers randomly to either the treatment or control groups usually using age, gender and disease stage variables to make sure the groups are of similar composition. Each patient receives a code number that matches the same code in his/her corresponding medication. When the patient doesn’t know whether they are getting treatment or control/placebo treatment, the trial is blind; in a double-blind trial the researchers/doctors are also unaware of which treatment is being administered to patients.
The final phase (4) starts immediately after the results of phase 3 trials are reviewed and considered to show good enough safety and efficacy to grant FDA approval; it consists of post-marketing monitoring over a longer period of time in a “real” scenario and wider population that may allow detection of rare side effects missed in earlier trials. This surveillance or “pharmacovigilance” may result in harmful effects reported over time, with consequences such as drug’s market withdrawal (or restricted use); this happened to troglitazone (Rezulin/Romozin) in 2000 due to risk of hepatotoxicity, cerivastatin (Baycol/Lipobay) in 2001 due to risk of rhabdomyolysis, and rofecoxib (Vioxx) in 2004 due to risk of myocardial infarction.
When pregnant women in other countries in Europe and Australia used the drug thalidomide in the 1960s for morning sickness, the drug was not marketed in the US thanks to a FDA employee (Dr. Kelsey, a woman) who felt there was no reliable safety evidence. When babies were born with severe birth defects such as hands and feet protruding from shoulders and hips and these were linked to thalidomide use, it prompted the 1962 Drug Amendments bill that changed drug regulation; it also resulted in the FDA excluding women from participating in clinical trials in 1977, which was reversed in 1993 although women are still a minority population in clinical trials. Dr. Kelsey's role in averting a thalidomide tragedy in the US was recognized with several awards in the US and abroad.
The final phase (4) starts immediately after the results of phase 3 trials are reviewed and considered to show good enough safety and efficacy to grant FDA approval; it consists of post-marketing monitoring over a longer period of time in a “real” scenario and wider population that may allow detection of rare side effects missed in earlier trials. This surveillance or “pharmacovigilance” may result in harmful effects reported over time, with consequences such as drug’s market withdrawal (or restricted use); this happened to troglitazone (Rezulin/Romozin) in 2000 due to risk of hepatotoxicity, cerivastatin (Baycol/Lipobay) in 2001 due to risk of rhabdomyolysis, and rofecoxib (Vioxx) in 2004 due to risk of myocardial infarction.
When pregnant women in other countries in Europe and Australia used the drug thalidomide in the 1960s for morning sickness, the drug was not marketed in the US thanks to a FDA employee (Dr. Kelsey, a woman) who felt there was no reliable safety evidence. When babies were born with severe birth defects such as hands and feet protruding from shoulders and hips and these were linked to thalidomide use, it prompted the 1962 Drug Amendments bill that changed drug regulation; it also resulted in the FDA excluding women from participating in clinical trials in 1977, which was reversed in 1993 although women are still a minority population in clinical trials. Dr. Kelsey's role in averting a thalidomide tragedy in the US was recognized with several awards in the US and abroad.

Children are another group, due to safety and ethical considerations, not usually included in clinical trials. For them to participate, a clinical benefit justifies the risk of taking the drug and they should be patients with the target disease or condition. Only about 50% of FDA approved drugs are labeled for use in children to date. However, clinical trials are common practice in pediatric cancer treatment, leading to considerable numbers of children with cancer participating in clinical trials.
This FDA web page has essential information and nice short videos explaining simply what clinical trials and phases are:
https://www.fda.gov/forpatients/approvals/drugs/ucm405622.htm
This FDA web page has essential information and nice short videos explaining simply what clinical trials and phases are:
https://www.fda.gov/forpatients/approvals/drugs/ucm405622.htm

 RSS Feed
RSS Feed